Parapareza spastyczna uwarunkowana mutacją BSCL2
Co-segregation of potentially benign variant of BSCL2 gene (rs145649423) with spastic paraplegia phenotype in Polish family
Łukasz Kępczyński†1,2, Katarzyna Połatyńska3, Mariusz Stasiołek4, Małgorzata Zielińska4, Przemysław Bojczuk4, Karol Jastrzębski5, Agata Michalska6, Mariusz Grudniak7, Dominika Szewczyk8, Tadeusz Kałużewski8, Anna Wójcicka9, Agnieszka Gach1
1Department of Genetics, Polish Mothers’ Memorial Institute Research Hospital (Łódź, Poland); 2Laboratory of Molecular Biology, Department of Internal Medicine, Kidney and Diabetes, Medical University of Łódź (Łódź, Poland); 3Department of Neurology, Polish Mothers’ Memorial Institute Research Hospital (Łódź, Poland); 4Department of Neurology, Medical University of Łódź (Łódź, Poland); 5Department of Neurology and Strokes, Medical University of Łódź (Łódź, Poland); 6Department of Physiotherapy, Collegium Medicum, Jan Kochanowski University (Kielce, Poland); 7Institute IurisMed – Independent Medical Examiners (Kutno, Poland); 8Laboratory of Medical Genetics of the “Genos” Partnership R&D Division (Łódź, Poland); 9Warsaw Genomics (Warsaw, Poland).
1. Introduction
Hereditary spastic paraplegias (HSPs) is an extremally heterogenous (phenotypic series in Online Mendlian Inheritance in Men include 71 entries) group of disorders of autosomal dominant (AD), autosomal recessive (AR) and X-linked (XL), mitochondrial and more complicated modes of inheritance on the border of dominant and recessive inheritance with most of the cases being sporadic (Boutry et al., 2019). HSPs exhibit significant clinical overlap with other hereditary disorders of nervous system as hereditary motor and sensory neuropathy (HMSN, Charcot-Marie-Tooth disease, CMT) and distal hereditary motor neuropathy. Overlapping phenotypes and heterogeneity of HSP lead to inefficiency of single-gene concurrent testing approach, and promotes more cost- and time-effective testing by multi-gene Next Generation Sequencing (NGS) panel approach.
Homozygous mutations in Berardinelli-Seip congenital dystrophy 2 gene (BSCL2) were attributed to generalized dystrophy syndrome originally described by Berardinelli, and confirmed by Seip (Lightbourne and Brown, 2017; Craveiro Sarmento et al., 2019). Heterozygous loss-of-function mutations in aforementioned gene, are associated with broad spectrum of neurological disorders including HSP, HMSN and distal hereditary motor neuropathy (Ito, 2005).
In this report we present the clear co-segregation of BSCL2 variant of conflicting interpretation, but generally treated as likely benign (rs145649423), with HSP phenotype with apparent AD mode of inheritance in one Polish family.
2. Material and methods
2.1. Patients
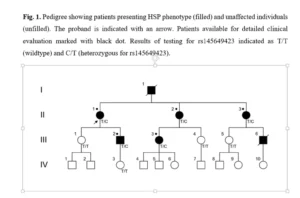
A Polish family was evaluated due to family history of HSP phenotype. The proband (patient II.1 in Fig. 1, indicated with an arrow) and 4 family members, presenting HSP phenotype (patients II.2, II.3, III.2, and III.3, indicated with black dot) were subject of detailed clinical evaluation. 9 family members (proband, spastic HSP presenting relatives, and their adult first degree descendants, not presenting HSP phenotype – individuals III.1, III.4, III.5, and IV.3) were available for genetic testing. All family members included in this study gave their informed consent to participate. All of the adult patients, both symptomatic and asymptomatic were examined by neurologist, children – descendants of affected parent were examined by child neurologist.
2.2. DNA analysis
Genomic DNA was obtained from peripheral blood leukocytes of the proband and indicated members of probands family using the Applied Biosystems 3130 Genetic Analyzer. In proband all coding exons with 10 – 20 nucleotide intronic flanks of following genes were analyzed: ATL1, BSCL2, HSPD1, KIAA0196, KIF5A, NIPA1, REEP1, RTN2, SLC33A1, SPAST, ZFYVE27. Amplicons were analyzed on NextSeq500 sequencer (Illumina). Variants were identified by Burrows-Wheeler Aligner. In the remaining family members, only the region of BSCL2 gene containing the locus of rs145649423 was analyzed. Primers for amplification of BSCL2 region (BSCL2_rs145649423_F: TGTCCGAGGAGGAGAAACCAGAT and BSCL2_rs145649423_R: GTGGGAAAGTGCTGGAATGT) were purchased from Merck.
3. Results and discussion
The proband – a 64 year-of-age female was referred to the outpatient genetic clinic due to progressive gait disturbance and paresthesias of late onset (around 40 year od age) with more severe exacerbation for 5 years without clear provoking agent. Previous examination included normal cerebrospinal fluid analysis, the patient was negative for Treponema pallidum, Borrelia testing showed IgM elevation, but the neuroboreliosis was excluded by Western-blot analysis, performed in another center. No neurocognitive decline was reported in neuropsychological examination. The family history was positive for gait disturbances of late onset – similar symptoms were also present in father (individual I.1, deceased), and probably in paternal grandfather (not shown on the pedigree). Gait disturbances were also present in two younger sisters of the proband (individuals II.2 and II.3), and their descendants – see below. The initial observation was consistent with hereditary spastic paraplegia with AD mode of inheritance. The proband and her two sisters were referred do Neurology Department for detailed clinical evaluation. High-throughput NGS testing for mutation in genes associated with AD-HSP was performed for the proband, and the DNA form peripheral blood leukocytes was archived for subsequent segregation analysis.
Testing of coding sequence and 10-20 nucleotide intron flanking sequences of 10 genes associated with uncomplicated and complicated with neuropathy AD-HSP ATL1 (SPG3A), HSPD1 (SPG13), KIF5A (SPG10), NIPA1 (SPG6), REEP1 (SPG31), RTN2 (SPG12), SLC33A1 (SPG42), SPAST (SPG4), WASHC5 (KIAA0166, SPG8), and ZFYVE27 (SPG33) revealed no variants classified as pathogenic, potentially non-pathogenic and of uncertain significance. BSCL2 gene analysis revealed presence of variant of conflicting interpretation: rs145649423 – NM_001122955.3:c.1280T>C (p.Leu427Pro) in heterozygous state. The presence of the T>C substitution was confirmed by Sanger sequencing. This variant is reported in ClinVar database as benign, likely benign and as of uncertain significance. The minor allele frequency of this variant is estimated as follows: The Genome Aggregation Database (gnomAD) – 0.00352, Exome Aggregation Consortium (ExAC) – 0.00392, The Genome Aggregation Database (gnomAD) – 0.00293, 1000 Genomes Project – 0.00080, NHLBI Exome Sequencing Project (ESP) Exome Variant Server – 0.00261, Trans-Omics for Precision Medicine (TOPMed) 0.00303 (data from ClinVar database). This variant was also reported as present in individuals with familial amyotrophic lateral sclerosis (FALS) (Morgan et al., 2015). Due to ongoing clinical evaluation and conflicting interpretation of variant pathogenicity, segregation analysis in siblings and descendants were performed.
Further clinical evaluation of the proband showed in magnetic resonance imaging non-specific, benign vascular lesions, and electrophysiological studies revealed axonal lesions of sensory nerves. Both sisters of the proband (II.2 and II.3) presented gait disturbances of similar age of onset, no signs of neuropathy in electrophysiological studies, and both inherited rs145649423 in heterozygous form. In the generation of descendants of proband and proband’s siblings, 3 of 6 individuals presented gait disturbances, and 5 of 6 individuals were available for molecular analysis (Fig. 2.). One of the affected individuals (III.6) was deceased due to hemorrhagic stroke, and was unavailable for genetic testing. Both individuals presenting HSP phenotype (III.2 and III.3) inherited rs145649423 in heterozygous form, whereas three unaffected individuals (III.1, III.4, and III.5) inherited wildtype form. None of the descendants from the IV generation presented HSP phenotype, nevertheless, none of children of affected parent, exceeded the age of onset in their relatives.
Heterozygous BSCL2 mutations are associated with vast variety of neurodegenerative disorders including uncomplicated and complicated HSP (Silver syndrome), HMSN/CMT disease, and FALS, described together as motor seipinopathy. Some reported families affected with pathogenic mutation in BSCL2 exhibited a wide range of clinical heterogeneity from rapid progressive neurodegenerative process to clinically asymptomatic individuals in the same family. Vast majority of BSCL2-dependant cases of neurogenerative disorders originate from two heterozygous gain-of-function mutations – rs137852972 – NM_001122955.3:c.455A>G (p.Asn152Ser) and rs137852973 – NM_001122955.3:c.461C>T (p.Ser154Leu) (Mussachio et al., 2017).
To our best knowledge, this is the first report of co-segregation of familial HSP phenotype with homogenous presentation regarding clinical picture of gait abnormalities and age of onset with rs1456494423 variant of BSCL2 gene. This clear co-segregation stays in contrast with bioinformatic interpretation of aforementioned variant as benign, likely benign or unknown significance. Co-segregation analysis is treated as a tool for clinical verification of variant of conflicting interpretation; in presented family, this co-segregation raised even more question than answers, especially regarding preclinical prognosis for children from IV generation. Further work is needed for full description of clinical significance of presented variant in population, regarding association with late-onset neurodegenerative disorders as well as its risk factor potential.

Opublikowano: : Nature Genetics
4. References
Boutry M, Morais S, Stevanin G. 2019. Update on the Genetics of Spastic Paraplegias. Curr Neurol Neurosci Rep. 19(4):18.
Craveiro Sarmento AS, Ferreira LC, Lima JG, de Azevedo Medeiros LB, Barbosa Cunha PT, Agnez-Lima LF, Galvão Ururahy MA, de Melo Campos JTA. 2019. The worldwide mutational landscape of Berardinelli-Seip congenital lipodystrophy. Mutat Res. 781:30-52.
Ito D. 2005 (update 2018). BSCL2-Related Neurologic Disorders/Seipinopathy. in: In: Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJH, Stephens K, Amemiya A, (ed.). 1993 – 2020. GeneReviews [Internet]. https://www.ncbi.nlm.nih.gov/books/NBK1307/
Lightbourne M, Brown RJ. 2017. Genetics of Lipodystrophy. Endocrinol Metab Clin North Am. 46(2):539-554.
Morgan S, Shoai M, Fratta P, Sidle K, Orrell R, Sweeney MG, Shatunov A, Sproviero W, Jones A, Al-Chalabi A, Malaspina A, Houlden H, Hardy J, Pittman A. 2015. Investigation of next-generation sequencing technologies as a diagnostic tool for amyotrophic lateral sclerosis. Neurobiol Aging. 36(3):1600.e5-8.
Musacchio T, Zaum AK, Üçeyler N, Sommer C, Pfeifroth N, Reiners K, Kunstmann E, Volkmann J, Rost S, Klebe S. 2017. ALS and MMN mimics in patients with BSCL2 mutations: the expanding clinical spectrum of SPG17 hereditary spastic paraplegia. J Neurol. 264(1):11-20.